

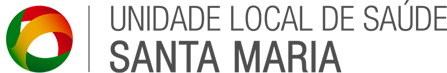
O que é a Polineuropatia Amiloidótica Familiar?
A amiloidose hereditária mediada por transtirretina (amiloidose ATTRv) é uma doença multissistémica, degenerativa associada a um largo espetro de manifestações clínicas, incluindo neuropatia sensitivo-motora e autonómica, cardiomiopatia, alterações gastrointestinais, nefropatia, envolvimento ocular e leptomeníngeo.
A forma neuropática (amiloidose ATTRv-PN), descrita em Portugal por Corino de Andrade em 19523, como uma forma de polineuropatia familiar é comummente conhecida, por Paramiloidose Familiar (PAF) ou Doença dos Pézinhos.
A Paramiloidose é uma doença rara, do adulto, crónica e progressiva que se manifesta habitualmente entre os 25 e os 40 anos, embora possa ocorrer depois dos 50 anos de idade, podendo ser fatal em 10 a 15 anos se não tratada.
Quais as causas?
Esta doença hereditária, que resulta de uma mutação (erro genético) no gene da transtirretina (TTR), é transmitida à geração seguinte de forma autossómica dominante, o que significa que 50% dos filhos de um doente poderão vir a herdar o gene responsável pela doença.
A doença é transmitida aos filhos por um dos pais biológicos, mesmo que este não apresente sinais ou sintomas da doença.
Por isso é muito importante falar com os seus familiares sobre a doença, para melhor perceber se há mais familiares afetados ou em risco de desenvolver a doença.
Qual a prevalência em Portugal?
Estima-se que mundialmente existam cerca de 10 000 doentes com amiloidose ATTRv-PN, na sua maioria associada à mutação Val30Met, dos quais cerca de 20%, em Portugal.
O maior foco da doença é descrito em Portugal (zona da Póvoa do Varzim/Vila do Conde) com uma prevalência bruta estimada de 22,93/100 000 habitantes, comparativamente ao resto do mundo (1/100 000). Calcula-se que em Portugal existam cerca de 71 novos casos /ano, oriundos de mais de 1200 famílias nucleares, estimando-se que existam atualmente 1865 indivíduos com ATTRv-PN em Portugal (45,8% homens; idade média 52,3 anos. A proporção de casos de início tardio (idade ≥50 anos), no total de casos incidentes, é de 28,7%, tendo-se registado um aumento deste tipo de casos, nos últimos anos.
Quais os sintomas da doença?
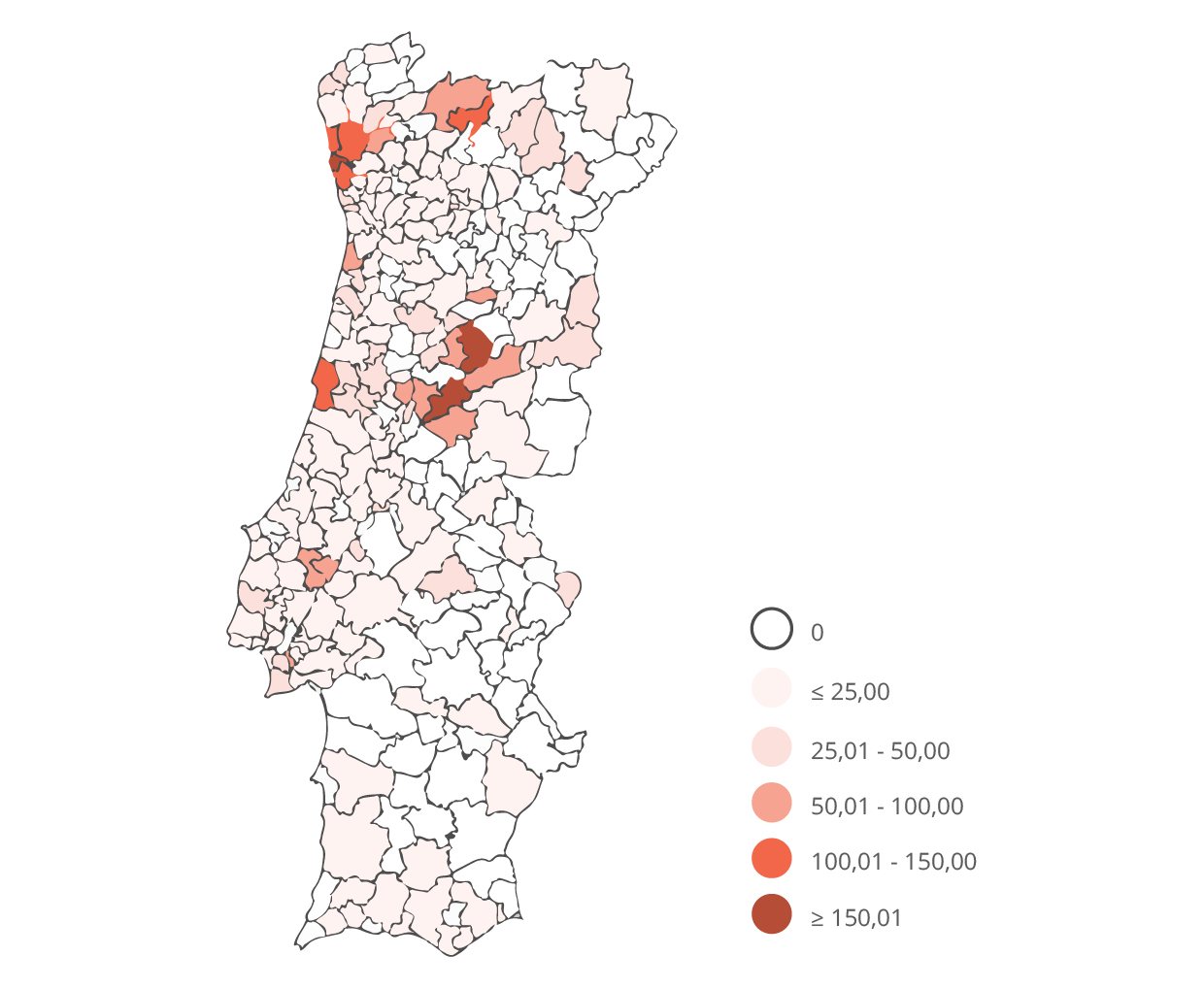
Os sintomas sensitivos são habitualmente os primeiros a surgir, em particular formigueiros, dor tipo choque elétrico, picada, ardor ou calor; diminuição da sensibilidade à temperatura (frio/quente), que começa nos pés e tem progressão ascendente, atingindo as mãos em cerca de 4 a 5 anos após o seu início. Com a progressão da doença, ocorre diminuição da força muscular que começa nos pés e pernas e progride para os membros superiores em anos, condicionando dificuldades motoras progressivas.
Noutros casos, a manifestação inicial da doença pode ser perda de peso involuntária, alterações do trânsito intestinal (obstipação ou diarreia alternando com obstipação), dificuldade em fazer as digestões ou um quadro de disfunção sexual. As alterações gastrointestinais vão-se tornando mais acentuadas, podendo associar-se náuseas e vómitos. Podem ocorrer alterações urinárias caracterizadas por dificuldade em esvaziar a bexiga completamente que se pode manifestar como infeções urinárias de repetição, evoluindo para incontinência urinária.
O envolvimento do coração pode não provocar sintomas numa fase inicial, ou manifestar-se como tonturas com as mudanças de posição, palpitações por alterações da condução e ritmo cardíaco e eventualmente pode ser necessário a colocação de pacemaker. Outra forma de manifestação cardíaca é a miocardiopatia por infiltração de substância amiloide, que pode levar a sintomas de insuficiência cardíaca.
O envolvimento renal pode ocorrer em cerca 30% dos doentes, e embora possa ser uma manifestação precoce da doença habitualmente surge numa fase tardia, conduzindo à falência do rim e à necessidade de diálise.
Podem também ocorrer perturbações visuais, como visão turva, olho seco, glaucoma ou diminuição da acuidade visual, caracteristicamente nas fases mais avançadas da doença.
Em fases mais avançadas da doença podem ocorrer episódios neurológicos transitórios mimetizando Acidentes Vasculares Cerebrais transitórios; crises epiléticas ou alterações progressivas de memória.
Sem tratamento, os sintomas agravam-se, causando a morte em média 10-15 anos após o seu aparecimento.
Como se diagnostica a paramiloidose?
O diagnóstico é realizado por teste genético (análise ao sangue) para confirmação da presença da mutação responsável pela doença. Pode ser necessário confirmar a presença de substância amiloide TTR em algum tecido através de biopsia das glândulas salivares ou gordura abdominal ou através de Cintigrafia cardíaca (DPD).
Nas famílias das pessoas afetadas, é possível identificar parentes portadores do gene alterado e que, por isso, estão em risco de desenvolver a doença. O teste genético está unicamente disponível para adultos (maiores de 18 anos) depois de estes terem recebido aconselhamento genético.
Uma vez que esta é uma doença de agravamento progressivo, é importante o diagnóstico precoce para que possa ser instituída terapêutica atempadamente.
Como comunicar o diagnóstico à família?
Na família das pessoas afetadas, é possível que existam familiares portadores do gene alterado e que, por isso, poderão vir a desenvolver a doença.
O teste genético pode ser feito nos familiares em risco (só após os 18 anos) mesmo que ainda não tenham aparecido sintomas da doença.
Deve incentivar os seus familiares a falar com os centros especializados de Paramiloidose, acerca do risco de terem herdado a doença.
Ao conhecer a doença também permitirá obter ajuda mais cedo, caso já apresentem alguns sintomas da doença.
Como se trata a paramiloidose?
Os doentes com paramiloidose devem ser acompanhados por uma equipa multidisciplinar envolvendo várias especialidades nomeadamente neurologia, genética médica, cardiologia, nefrologia, oftalmologia entre outras, de acordo com o envolvimento de outros órgãos que possam surgir no decurso da doença.
O tratamento modificador da doença deve ser iniciado o mais precocemente possível e em centros especializados. Existem atualmente diferentes tratamentos modificadores para a doença como o transplante hepático, Tafamidis (comprimido oral), Inotersen e Patisiran (injetáveis).
A escolha do tratamento mais adequado depende de vários fatores clínicos como a idade, gravidade da doença e presença de comorbilidades. Existem critérios bem definidos a ser discutidos em consulta especializada.
Para lá destes tratamentos, importa referir que os sinais e sintomas da paramiloidose são variados e os pacientes podem necessitar de outros tratamentos sintomáticos para manter as suas atividades diárias.
Atividades do CR
O CR de Referencia de Paramiloidose do CHULN tem um conjunto de atividades assistenciais destinadas a:
- Diagnóstico de Amiloidose Hereditária relacionada com a transtirretina (Paramiloidose) na Consulta de Paramiloidose/Neurologia e restantes consultas pertencentes ao CR de Paramiloidose do CHULN;
- Abordagem terapêutica multidisciplinar dos doentes com Paramiloidose diagnosticados de novo ou em seguimento no CHULN;
- Seguimento em ambulatório, sequencial de ajuste e monitorização terapêutica.
Localização e Contactos do CR
Torre de Neurociências, Elevador 1 Piso 7
Telef: 217805219
Email: cr.paramiloidose@chln.min-saude.pt
